
- 제약·바이오
- 국내사
제2 발사르탄 사태없다…식약처, 의약품제조소 특별점검

발행날짜: 2019-01-28 11:06:57
-
가
-
 URL복사
URL복사 -
-




2019년 중점 추진 사업 공개 "원료부터 제조·유통까지 의약품 안전 관리 강화"
식품의약품안전처가 발사르탄 오염물 혼입 사태를 맞아 올해 중점 추진 계획으로 의약품의 안전 관리에 방점을 찍었다.
식약처는 원료 해외제조소 사전 등록제부터 의약품 품질 고도화시스템(QbD: Quality by Design) 등 글로벌 수준의 안전제도를 구축하고 2월부터 특별점검단을 구성해 원료의약품 제조소와 수입업체를 집중 점검한다는 계획이다.
28일 식약처 류영진 처장은 업무계획 브리핑을 통해 2019년 역점 추진 과제를 공개했다.
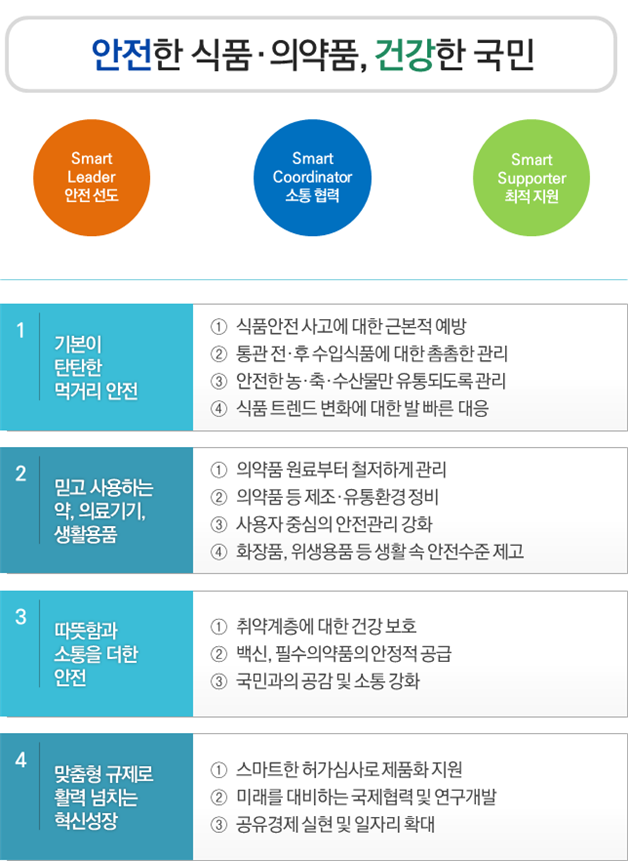
중점 업무 추진 방향은 ▲기본이 탄탄한 먹거리 안전 ▲믿고 사용하는 약·의료기기·생활용품 ▲따뜻함과 소통을 더한 안전 ▲맞춤형 규제로 활력 넘치는 혁신성장까지 주로 '안전'에 무게를 뒀다.
식약처는 의약품 제조공정 중에 불순물이 생성돼 문제가 된 고혈압약(발사르탄) 사건의 재발 방지를 위해 허가‧등록 시 제조업체로 해금 불순물 등 유해물질에 대한 안전성 자료를 제출토록 의무화(3월) 하는 등 원료의약품 관리를 강화한다.
또 해외에서 원료의약품을 수입해 오는 경우에도 해외 제조업소를 사전에 등록하도록 의무화(12월)하고, 위험성이 높은 제조업소를 우선순위로 해 현지실사를 확대키로 했다.
식약처는 특별점검단을 구성해 국내 원료의약품 제조소와 수입업체에 대해 품질관리 적정성 등을 2월과 3월에 걸쳐 집중 점검한다.
9월 대한민국 약전의 전면 개정도 이뤄진다.
이를 통해 안전기준을 국제기준에 맞게 대폭 신설·강화하고, 의약품 품질고도화시스템(QbD : Quality by Design)을 제조품질관리기준(GMP)에 반영한다는 계획이다.
식약처는 "제네릭, 의료기기 안전성·유효성 검증을 강화한다"며 "유통 의약품의 안전과 품질관리를 강화하기 위해 2월부터 제네릭 허가제도를 개선하고, 국제수준에 부합하는 심사자료 제출 의무화를 추진하겠다"고 강조했다.
이어 "8월부터 인체 이식형 의료기기는 이미 출시된 의료기기와의 동등성 인정여부와 관계없이 반드시 임상시험을 통해 안전성과 유효성을 검증하도록 의무화하겠다"며 "의약품 허가갱신 시 보험청구 실적까지 반영해 실제 사용되지 않은 제품은 시장퇴출 추진한다"고 밝혔다.
이외 의료기기도 주기적으로 안전성‧유효성을 재검토하는 품목 허가갱신 제도도 11월 도입된다.
사용자 중심의 안전관리를 위해 식약처는 의약품 부작용 피해에 대한 국가 보상범위를 비급여 진료비까지 확대하고, 임상시험에 대해서도 표준 피해보상 절차를 6월까지 마련한다.
식약처는 "10월에는 환자 신고, 병원 의무기록 등 실제 의료현장의 부작용 정보에 대한 통합 분석체계 마련하겠다"며 "안전사용 정보(DUR)를 반영하지 않은 처방의 부작용 모니터링 등 능동적으로 정보를 수집하고 이를 공개하겠다"고 덧붙였다.
한약재 벤조피렌 등 유해물질을 조사(1∼6월)하고, 주사제 등 액체 형태 의약품의 용기·포장에서 나오는 유해물질 관리방안 마련(9월), 마약류 통합관리 시스템의 빅데이터 분석을 통해 오남용이 의심되는 마약류 유통·취급자 선별 및 감시한다.
각 의사에게 마약류 처방내역 비교·분석결과 제공(3월)하고 환자가 본인의 마약류 투약내역을 확인할 수 있는 시스템은 9월부터 개발에 돌입한다.
한편 희귀 의약품, 긴급도입 필요 의료기기 등에 대한 공적 공급 확대된다.
3월부터 희귀·난치질환자가 필요로 하는 해외 대마성분 의약품에 대해 한국희귀·필수의약품센터를 통한 수입을 허용하고, 어린이용 인공혈관 등 희소‧긴급도입 필요 의료기기는 6월부터 국가가 우선 비용을 지원해 신속 공급하는 제도를 도입한다.
5월부터 국내 임상시험 의약품뿐만 아니라 해외 임상 의약품도 환자치료 목적 사용을 허용하고, 사용승인 기간도 대폭 단축(7일→즉시)한다.
식약처는 원료 해외제조소 사전 등록제부터 의약품 품질 고도화시스템(QbD: Quality by Design) 등 글로벌 수준의 안전제도를 구축하고 2월부터 특별점검단을 구성해 원료의약품 제조소와 수입업체를 집중 점검한다는 계획이다.
28일 식약처 류영진 처장은 업무계획 브리핑을 통해 2019년 역점 추진 과제를 공개했다.
중점 업무 추진 방향은 ▲기본이 탄탄한 먹거리 안전 ▲믿고 사용하는 약·의료기기·생활용품 ▲따뜻함과 소통을 더한 안전 ▲맞춤형 규제로 활력 넘치는 혁신성장까지 주로 '안전'에 무게를 뒀다.
또 해외에서 원료의약품을 수입해 오는 경우에도 해외 제조업소를 사전에 등록하도록 의무화(12월)하고, 위험성이 높은 제조업소를 우선순위로 해 현지실사를 확대키로 했다.
식약처는 특별점검단을 구성해 국내 원료의약품 제조소와 수입업체에 대해 품질관리 적정성 등을 2월과 3월에 걸쳐 집중 점검한다.
9월 대한민국 약전의 전면 개정도 이뤄진다.
이를 통해 안전기준을 국제기준에 맞게 대폭 신설·강화하고, 의약품 품질고도화시스템(QbD : Quality by Design)을 제조품질관리기준(GMP)에 반영한다는 계획이다.
식약처는 "제네릭, 의료기기 안전성·유효성 검증을 강화한다"며 "유통 의약품의 안전과 품질관리를 강화하기 위해 2월부터 제네릭 허가제도를 개선하고, 국제수준에 부합하는 심사자료 제출 의무화를 추진하겠다"고 강조했다.
이어 "8월부터 인체 이식형 의료기기는 이미 출시된 의료기기와의 동등성 인정여부와 관계없이 반드시 임상시험을 통해 안전성과 유효성을 검증하도록 의무화하겠다"며 "의약품 허가갱신 시 보험청구 실적까지 반영해 실제 사용되지 않은 제품은 시장퇴출 추진한다"고 밝혔다.
이외 의료기기도 주기적으로 안전성‧유효성을 재검토하는 품목 허가갱신 제도도 11월 도입된다.
사용자 중심의 안전관리를 위해 식약처는 의약품 부작용 피해에 대한 국가 보상범위를 비급여 진료비까지 확대하고, 임상시험에 대해서도 표준 피해보상 절차를 6월까지 마련한다.
식약처는 "10월에는 환자 신고, 병원 의무기록 등 실제 의료현장의 부작용 정보에 대한 통합 분석체계 마련하겠다"며 "안전사용 정보(DUR)를 반영하지 않은 처방의 부작용 모니터링 등 능동적으로 정보를 수집하고 이를 공개하겠다"고 덧붙였다.
한약재 벤조피렌 등 유해물질을 조사(1∼6월)하고, 주사제 등 액체 형태 의약품의 용기·포장에서 나오는 유해물질 관리방안 마련(9월), 마약류 통합관리 시스템의 빅데이터 분석을 통해 오남용이 의심되는 마약류 유통·취급자 선별 및 감시한다.
각 의사에게 마약류 처방내역 비교·분석결과 제공(3월)하고 환자가 본인의 마약류 투약내역을 확인할 수 있는 시스템은 9월부터 개발에 돌입한다.
한편 희귀 의약품, 긴급도입 필요 의료기기 등에 대한 공적 공급 확대된다.
3월부터 희귀·난치질환자가 필요로 하는 해외 대마성분 의약품에 대해 한국희귀·필수의약품센터를 통한 수입을 허용하고, 어린이용 인공혈관 등 희소‧긴급도입 필요 의료기기는 6월부터 국가가 우선 비용을 지원해 신속 공급하는 제도를 도입한다.
5월부터 국내 임상시험 의약품뿐만 아니라 해외 임상 의약품도 환자치료 목적 사용을 허용하고, 사용승인 기간도 대폭 단축(7일→즉시)한다.