



FDA 세포․유전자치료제 심사인력 100명 확충 계획
국내 신기술 수요 증가 따른 심사인력 증가 목소리
신기술의 등장에 따른 세포 및 유전자치료제의 등장이 늘어나면서 이를 심사하기 위해 미국 식품의약국(이하 FDA)이 신약허가 심사인력을 대폭 늘린다.

지난 26일 FDA 바이오의약품평가연구센터(CBER) 조직 및 첨단치료제과(Office of Tissues and Advanced Therapies) 윌슨 브라인언 과장은 미국 세포유전자학회 정책회담에서 현재 세포 및 유전자치료제 관련 3000개 이상의 임상시험계획(IND)을 처리해야 한다고 밝혔다.
윌슨 과장에 따르면 지난 5년~10년 동안 IND 신청 건수와 상담 업무량이 FDA 심사자 및 프로젝트 관리자 증가를 훨씬 초과하는 속도로 증가한 상태.
과거 조직 및 첨단치료제과에 제출된 신규 IND 건수는 지난 2016년 163건에 불과했으나 ▲2020년 350건 ▲2021년 299건 등으로 급증했으며, 2022년에는 더 증가할 것으로 예상되고 있다.
아울러 윌슨 과장은 지난 5월 19일 개최된 미국 세포유전자학회에서 늘어나는 업무량을 해결하기 위해 가이던스, 워크숍, 웨비나 등을 통한 그룹 커뮤니케이션(group communication)의 방안을 검토하고 있다고 밝힌 바 있다.
FDA에서 운영하고 있는 혁신치료제(BreakThrough) 및 재생의료첨단치료제(RMAT) 지정 제도를 통한 신청도 늘고 있기 때문에 지정된 세포 및 유전자치료제를 우선 심사가 필요해 다른 IND 신청에 대한 검토가 우선순위에서 밀려나고 있다는 게 그의 설명이다.
이 같은 이유로 현재 FDA 조직 및 첨단치료제과에는 300명이 근무하고 있지만 향후 제7차 전문의약품 이용자부담금법(PDUFA Ⅶ)이 재승인 시 향후 4~5년에 걸쳐 약 100명의 직원을 추가로 채용될 예정이라고 밝혔다.
국내로 눈을 돌려보면 의약품, 의료기기 업계에서는 식품의약품안전처 심사인력을 늘려달라는 요구를 지속적으로 하고 있다.
특히, 세포․유전자치료제, 마이크로바이옴 등과 같은 새로운 치료제를 심사하기 위해서는 새로운 전문심사자를 채용해 신속히 심사하고, 업계와 소통하고, 필요한 경우 인허가 가이드라인을 제․개정하는 등의 선제적인 조치가 필요하다는 지적.
식약처 역시 인력확보와 가이드라인 마련을 위해 노력하고 있지만 FDA와 유럽의약품청(EMA)의 인력과 비교하면 그 수가 적은 상황이다.
여기에는 FDA와 같이 심사비용을 높여서 효율적인 심사과정을 구축해야 된다는 의견도 존재하지만 심사관 연봉 등이 여러 규정이 묶여있다는 점을 고려했을 때 즉각적인 인력확보로 연결될지 여부는 미지수다.
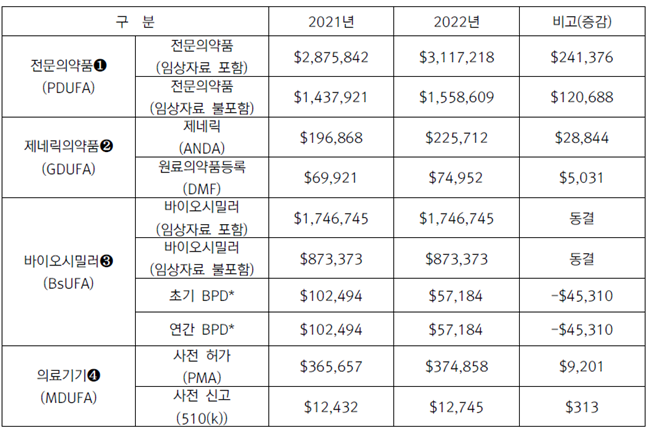
미국의 경우 전문의약품 이용자부담금법(Prescription Drug User Fee Act)을 통해 기업들이 FDA에 전문의약품의 시판 허가 등을 위해 내야 하는 심사수수료에 관한 법으로 1992년 제정했다.
이후 5년마다 미국 의회의 승인을 받고 있으며, 올해 10월부터 7번째 법의 적용을 받게 된다. 현재는 제네릭의약품, 바이오시밀러, 의료기기의 경우도 별도 이용자부담금법에 적용을 받고 있다.
제약업계 관계자는 "정부가 공무원 정원 감축 기조를 밀어붙이면서 식약처 심사 인력 감축 가능성이 커지고 있는데, 이는 글로벌 기조와 반대되는 부분"이라며 "제약바이오업계의 글로벌 진출을 위해서라도 심사에 대한 부분이 개선되기를 바란다"고 덧붙였다.