자디앙듀오 후발주자 경쟁력은 서방형 제제...국내 첫 허가
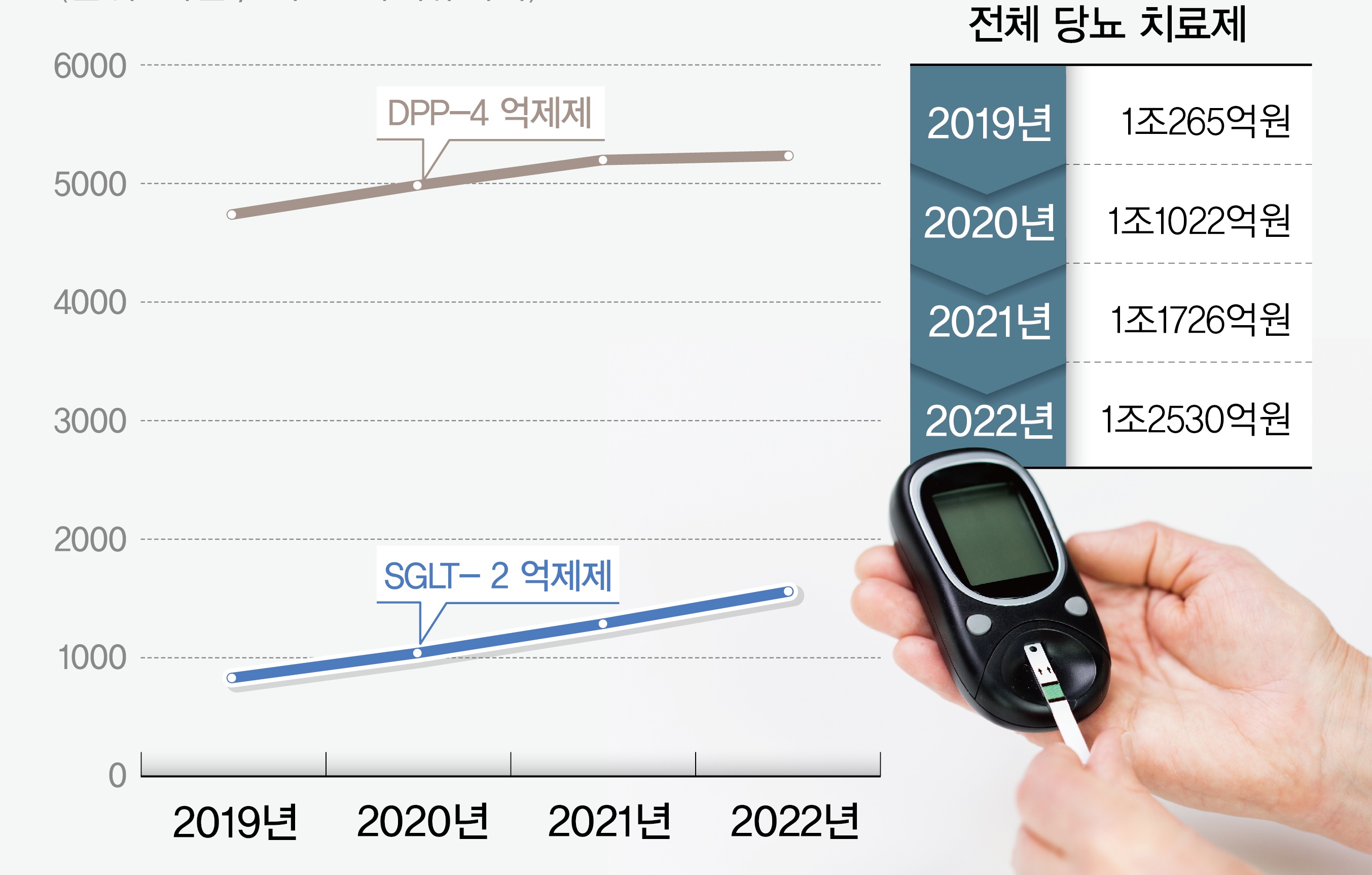
메디칼타임즈=허성규 기자자디앙과 자디앙듀오에 대한 국내사들의 제네릭 허가가 다수 이뤄진 가운데, 오리지널에도 없는 서방형 제제가 등장했다.이는 당뇨병치료제 시장이 포화 상태가 되면서 차별화 전략을 펼친 것으로, 이후에도 서방형 제제가 허가를 이어갈 것으로 예상된다.도전이 이어지고 있는 '자디앙듀오' 제품사진11일 동광제약은 식품의약품안전처로부터 ‘엠플로엠서방정10/1000mg’을 허가 받았다.해당 품목은 엠파글리플로진과 메트포르민 복합제제로 오리지널은 베링거인겔하임의 ‘자디앙듀오’다.자디앙듀오는 베링거인겔하임의 SGLT-2 억제제 계열의 자디앙과 메트포르민 복합제로 현재 국내사 다수가 관심을 가지는 품목이다.자디앙듀오의 경우 미등재 특허 등에 따라 2025년 10월 존속기간 만료 이후에나 출시가 가능하지만 이미 186개에 달하는 품목이 허가를 받은 상태다.여기에 특허 만료까지 기간이 남아있다는 점에서 추가적인 제네릭 허가 역시 가능한 상황.이처럼 특허 만료 이후 치열한 경쟁이 예고되는 만큼 제약사들은 차별화 전략으로 서방형 제제를 택했다.현재 자디앙듀오는 엠파글리플로진 5mg과 12mg에 메트포르민 500mg, 850mg, 1000mg의 조합으로 총 6개 품목이 허가된 상태다.즉 오리지널인 자디앙듀오도 갖추지 못한 서방형 제제를 통해 시장에서의 입지 확대를 노리는 것.이번에 처음으로 허가 받은 동광제약 외에도 이미 다른 제약사들 역시 개발에 착수했다는 점도 주목된다.실제로 이미 경보제약과 에이프로젠바이오로직스는 해당 조합의 서방형 제제에 대한 임상 1상을 4건 씩 이상 승인 받았다.결국 동광제약이 먼저 허가를 받으며 한발 나아갔지만, 다른 제약사들의 허가가 이어질 전망이다.아울러 허가를 획득하는 제약사들이 허여를 통해 위수탁 품목을 확대할 경우 품목은 더욱 늘어날 수 있다.한편 최근 이같은 서방형 제제 개발은 국내사들의 트렌드 중 하나다.앞서 DPP-4 억제제 계열의 당뇨병 치료제인 트라젠타듀오와 동일한 성분의 서방형 제제인 '트라리틴콤비서방정'을 대원제약이 허가 받기도 했다.